|
Geometrical descriptors
|   |
|
Geometrical descriptors
| |
List of geometrical descriptors calculated by DRAGON
Geometrical descriptors are defined in several different ways but always derived from the three-dimensional structure of the molecule. Generally, geometrical descriptors are calculated either on some optimised molecular geometry obtained by the methods of the computational chemistry or on crystallographic coordinates.
Since a geometrical representation of a molecule involves the knowledge of the relative positions of the atoms in 3D space, i. e., the (x,y,z) atomic coordinates of the molecule atoms, geometrical descriptors usually provide more information and discrimination power also for similar molecular structures and molecule conformations than topological descriptors. Despite their high information content, geometrical descriptors usually show some drawbacks. They require geometry optimisation and therefore the overhead to calculate them. Moreover, for flexible molecules, several molecule conformations can be available: on one hand, new information is available and can be exploited, but, on the other hand, the problem complexity can significantly increase.
Many molecular descriptors calculated by DRAGON and belonging to the block of geometrical descriptors are commonly known as topographic indices, being calculated on the graph representation of molecules but using the geometric distances between atoms instead of the topological distances.
These are mainly derived from the geometry matrix which is a square symmetric matrix collecting the geometric distances (Euclidean distances) between all pairs of atoms. A row sum of the geometry matrix, called geometric distance degree, is the sum of the geometric distances from an atom to any other atom in the molecule.
The distance/distance matrix is a square symmetric matrix whose entries are the ratios of the geometric over topological distance between all pairs of atoms [M. Randic, A.F. Kleiner, L.M. DeAlba, J.Chem.Inf.Comput.Sci. 1994, 34, 277-286].
Gravitational indices are molecular descriptors reflecting the mass distribution in a molecule, defined as [A.R. Katritzky, L. Mu, V.S. Lobanov, M. Karelson, J.Phys.Chem. 1996, 100, 10400-10407]:
![]()

where mi and mj are the atomic masses of the considered atoms, rij the corresponding interatomic distances, nAT and nBT the number of atoms and bonds of the molecule, respectively. The G1 index takes into account all atom pairs in the molecule while the G2 index is restricted to pairs of bonded atoms. These indices are related to the bulk cohesiveness of the molecules accounting, simultaneously, for both atomic masses (volumes) and their distribution within the molecular space.
The radius of gyration (RGyr) is a size descriptor for the distribution of atomic masses in a molecule [G.A. Arteca, Molecular Shape Descriptors in Reviews in Computational Chemistry - Vol. 9, K.B. Lipkowitz, D. Boyd (Eds.), VCH Publishers, New York (NY), pp. 191-253, 1991], calculated as:
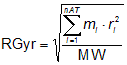
where ri is the distance of the ith atom from the centre of mass of the molecule, mi is the corresponding atomic mass, nAT the atom number and MW the molecular weight.
The span R (SPAN) is a size descriptor defined as the radius of the smallest sphere, centred on the centre of mass, completely enclosing all atoms of a molecule [G.A. Arteca, Molecular Shape Descriptors in Reviews in Computational Chemistry - Vol. 9, K.B. Lipkowitz, D. Boyd (Eds.), VCH Publishers, New York (NY), pp. 191-253, 1991]:
![]()
where ri is the distance of the ith atom from the centre of mass.
The average span R (SPAM) is the root square of the ratio of SPAN over the number of atoms.
The molecular eccentricity (MEcc) is a shape descriptor calculated from the eigenvalues l of the molecular inertia matrix [G.A. Arteca, Molecular Shape Descriptors in Reviews in Computational Chemistry - Vol. 9, K.B. Lipkowitz, D. Boyd (Eds.), VCH Publishers, New York (NY), pp. 191-253, 1991]:
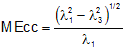
It ranges from 0 to 1, value 0 corresponding to spherical top molecules and value 1 to linear molecules.
The spherosity (SPH) is an anisometry descriptor calculated as a function of the eigenvalues of the covariance matrix calculated from the molecular matrix:
![]()
Spherosity index varies from zero for flat molecules, such as benzene, to one for totally spherical molecules [D.D. Robinson, T.W. Barlow, W.G. Richards, J.Chem.Inf.Comput.Sci. 1997, 37, 939-942].
The asphericity (ASP) is an anisometry descriptor which measures the deviation from the spherical shape [G.A. Arteca, Molecular Shape Descriptors in Reviews in Computational Chemistry - Vol. 9, K.B. Lipkowitz, D. Boyd (Eds.), VCH Publishers, New York (NY), pp. 191-253, 1991], calculated from the eigenvalues l of the molecular inertia matrix as follows:
![]()
Asphericity varies from 0 for spherical top molecules to 1 for linear molecules. For prolate molecules (cigar-shaped), l1 � l2 > l3 and ASP � 0.25, whereas for oblate molecules (disk-shaped), l1 > l2 � l3 and ASP � 1.
The length-to-breadth ratio is commonly the ratio of the longest L to the shortest B side of a rectangle containing some molecular projection, once univocally defined a specific molecular orientation. The length-to-breadth ratio by WHIM (L/Bw) is calculated as the ratio between the first and second eigenvalue of the molecular inertia matrix. It can be noted that this shape parameter not only accounts for the distance between extreme atoms along the principal axes but also for the distribution of all atoms around the molecule centre.
Aromaticity indices encode information on the electron delocalisation degree of a molecule [T.M. Krygowski, A. Ciesielski, C.W. Bird, A. Kotschy, J.Chem.Inf.Comput.Sci. 1995, 35, 203-210]. In spite of the concept of aromaticity has never been defined unequivocally, the commonly accepted description of aromaticity is as a characterictic delocalisation of the p-electrons giving a stabilisation of cyclic and polycyclic conjugated molecules, i.e. the increased stability of conjugated rings compared to its classical localised structure.
Aromaticity indices are often calculated from bond lengths and bond orders of the compounds; therefore, their values closely depend on the molecule optimisation procedure and the codification of conjugated bonds.

where the first sum runs over each conjugetd bond type, Bpk is the number of considered p-bond contributions of the kth conjugated bond type, rb is the actual bond length, ak and rkopt are a numerical constant and the typical conjugated bond length referring to the kth conjugated bond type (see values in the table below). Bp is the total number of bonds belonging to conjugated systems.
|
Bond |
a |
ropt |
Bond |
a |
ropt |
|
C � C (butadiene) |
257.7 |
1.388 |
C � P |
118.91 |
1.698 |
|
C � C |
98.89 |
1.397 |
C � S |
94.09 |
1.677 |
|
C � N |
93.52 |
1.334 |
N � N |
130.33 |
1.309 |
|
C � O |
157.38 |
1.265 |
N � O |
57.21 |
1.248 |
![]()
where Bp is the total number of conjugated bonds, the sum runs over each conjugated bond type, Bpk is the number of considered p-bond contributions of the kth conjugated bond type, rb is the actual bond length, ak and rkopt are a numerical constant and the typical aromatic bond length referring to the kth aromatic bond type (see values in the table above). This descriptor depends on the conjugation degree of a molecule as well as on the total number of p bonds.

where the sum runs over the bonds belonging to aromatic rings, ![]() is the p bond average length and rp are the actual p bond lengths, Bp is the number of aromatic bonds.
is the p bond average length and rp are the actual p bond lengths, Bp is the number of aromatic bonds.
Based on the same principles of the WHIMs, COMMA2 descriptors have recently been proposed [B.D.Silverman, J. Chem. Inf. Comput. Sci. 2000, 40, 1470-1476]. They consist of 11 descriptors given by moment expansions for which the zero-order moment of a property field is nonvanishing. DRAGON calculates 4 COMMA2 descriptors for 4 different molecular properties: mass (m), van der Waals volume (v), Sanderson electronegativity (e), polarizability (p). COMMA2 descriptors provided by DRAGON for each molecular property w are: